
AIport Syndrome
Overview of AIport Syndrome
Alport Syndrome (also known as hereditary nephritis) is a rare genetic disorder that results from mutations in the genes encoding alpha chains of type IV collagen, which is a component of the glomerular basement membrane (1). Type IV collagen alpha chains are primarily located in the kidneys, eyes, and cochlea (1). A reported gene mutation frequency of 1:5,000 is responsible for Alport Syndrome (3). The condition is predominantly X-linked and therefore more sever in males. However, it can also be autosomal recessive (ARAS) or autosomal dominant (ADAS) (Table 1)(1,2). Alport syndrome is not known to be associated with any race or geographic location (3).
Specifically, mutations in type IV collagen genes COL4A3, COL4A4, and/or COL4A5 lead to Alport Syndrome (1,2,3). Type IV collagen is formed by a triple helix of COL4A3, COL4A4, and COL4A5 and mutations in these genes result in a missing or deformed typical triple helix structure (4). This causes the glomerular basement membrane (GBM) to split and become lamellated, and podocyte effacement occurs, resulting in glomerulosclerosis and kidney fibrosis (4).
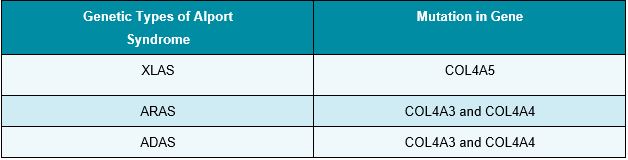
Table 1: Alport Syndrome Type and Mutation
XLAS is caused by mutations in the COL4A5 gene and is the most common form of Alport Syndrome (1,2). The X-linked (related to the X chromosome) variant of this disease affects about 80% of patients (5). Males with X-linked COL45 mutations experience more severe symptoms than females with same mutation(s) since females are XX and males as XY (6). By the age of 40, 90% of males without therapy experience kidney failure. Kidney failure strikes women less commonly and more gradually (5). ARAS and ADAS are caused by mutations in the COL4A3 and COL4A4 genes (1). ARAS occurs when both parents have recessive gene mutations that are independently passed to the offspring. In contrast, ADAS occurs as a dominant gene mutation (5). Possession of a single copy of the defective gene results in Thin Basement Membrane nephropathy.
Diagnosis
Signs and symptoms of Alport Syndrome other than kidney damage, eye abnormalities, and hearing loss include hematuria, proteinuria, hypertension, and edema in the lower extremities as well as around the eyes (1,2,3,5). Depending on the age, gender, and inherited form of Alport Syndrome, these indications and symptoms may vary (5). A diagnosis of Alport Syndrome can be obtained using a urine test, blood test, glomerular filtration rate (GFR), hearing test, and vision test. This diagnosis is then confirmed by a kidney biopsy showing lamellation of the GBM with electron microscopy and genetic testing to determine the haplotype or phenotype of the condition (5).
Treatment
There is currently no specific treatment or cure for Alport Syndrome. In order to limit the progression of proteinuria and kidney damage, management of existing symptoms is imperative. For the treatment of proteinuria, hypertension, and CKD, angiotensin- converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs) are considered first-line therapy while spironolactone is considered as second-line therapy (7). In patients with Alport Syndrome, ACE inhibitors and/or ARBs may postpone the need for dialysis and may reduce proteinuria and slow renal progression (1,7).
For ongoing management, patients should be assessed for proteinuria (urinary protein excretion of > 150 mg/day) annually with a urinalysis and obtain serum creatinine (normal: 0.7-1.3 mg/dL for men and 0.6-1.1 mg/dL for women) and blood pressure to monitor kidney function (2,8,9). Blood pressure should be measured to monitor kidney function considering damaged kidneys fail to regulate blood pressure (10). Patients’ hearing should also be assessed every 1-2 years for sensorineural hearing loss and a need for hearing aids (2). Clear lens phacoemulsification with intraocular lens implantation is a treatment option for patients with ocular involvement, particularly anterior lenticonus (1). For all members of the affected family, psychosocial support is crucial (11).
Although medications may delay the onset of kidney impairment, the majority of patients with Alport Syndrome will eventually need dialysis or a kidney transplant (1,5). Kidney transplants in patients with Alport Syndrome have consistently demonstrated the same or better survival compared to other causes of end-stage kidney disease (12). Additionally, kidney transplants do not result in the return of Alport Syndrome. However, approximately 3% of Alport transplant recipients produce antibodies against the alpha-3(IV) collagen chain in the transplanted kidney resulting in anti-GBM nephritis (6).
At this time, gene therapy and/or gene editing is not an available treatment option for Alport Syndrome; however, a few CRISPR/Cas9 gene therapy-based trials have begun over the last few years. As a result of structural defects that cause apoptosis and cell death, the number of podocytes steadily declines in patients with Alport Syndrome (13). Since a functional GBM can still theoretically be reconstituted during the early stages of the disease, CRISPR/Cas9 gene therapy is anticipated to be more effective during this time (13,14). Despite these encouraging findings, there is still a long way to go before this proof of concept can be transferred to in vivo tests because manipulating podocytes is challenging. The therapeutic approach with the best potential to reverse this condition is possibly gene therapy, which targets faulty collagen chains early in the disease (14).
References:
- Watson S, Padala SA, Bush Alport Syndrome. [Updated 2022 Aug 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470419/
- Kashtan CE. Alport Syndrome. 2001 Aug 28 [Updated 2019 Feb 21]. In: Adam MP, Everman DB, Mirzaa GM, et , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1207/
- Alves FR, de A Quintanilha Ribeiro F. Revision about hearing loss in the Alport’s syndrome, analyzing the clinical, genetic and bio-molecular Rev Bras Otorrinolaringol (Engl Ed). 2005 Nov-Dec;71(6):813-9.
- Kruegel J, Rubel D, Gross Alport syndrome–insights from basic and clinical research. Nat Rev Nephrol. 2013 Mar;9(3):170-8.
- Alport National Kidney Foundation. (2022, September 19). Retrieved October 26, 2022, from https://www.kidney.org/atoz/content/alport
- Alport NORD (National Organization for Rare Disorders). (2020, April 7). Retrieved October 31, 2022, from https://rarediseases.org/rare-diseases/alport- syndrome/#:~:text=Males%20have%20one%20X%20and,males%20than%20in%20affected%20females
- Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013 Feb;24(3):364-75.
- Carroll MF and Temte Proteinuria in Adults: A Diagnostic Approach. Am Fam Physician. 2000;62(6):1333-1340.
- Creatinine – serum. ucsfhealth.org. (2020, October 6). Retrieved October 30, 2022, from https://www.ucorg/medical-tests/creatinine-blood- test#:~:text=A%20normal%20result%20is%200.7,lower%20creatinine%20level%20than%20men.
- How high blood pressure can lead to kidney damage or failure. heart.org. (2022, March 4). Retrieved October 30, 2022, from https://www.heart.org/en/health-topics/high-blood-pressure/health-threats-from-high- blood-pressure/how-high-blood-pressure-can-lead-to-kidney-damage-or-failure
- Nicklason E, Mack H, Beltz J, Jacob J, Farahani M, Colville D, Savige Corneal endothelial cell abnormalities in X-linked Alport syndrome. Ophthalmic Genet. 2020 Feb;41(1):13-19.
- Kashtan CE. Renal transplantation in patients with Alport syndrome: patient selection, outcomes, and donor evaluation. Int J Nephrol Renovasc Dis. 2018;11:267-270. Published 2018 Oct 16. doi:10.2147/IJNRD.S150539
- Chavez E, Rodriguez J, Drexler Y, Fornoni Novel Therapies for Alport Syndrome. Front Med (Lausanne). 2022 Apr 25;9:848389.
- Daga S, Donati F, Capitani K, Croci K, Tita R, Giliberti A, et al.. New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur J Hum Genet. (2020) 28:480-90
GMO-000823 Rev A 03/2024